The following is adapted from a testimony given to the Senate Special Committee on Aging on October 8, 2025. Video of the hearing and full written testimony are available here. Please note that the full 15-page written testimony is attached as a download.
Introduction
Drugs, especially generic drugs, are a key component of medical care for seniors. Nearly nine in ten adults aged 65 and older use at least one prescription medication, and more than half take four or more on a regular basis, with the share increasing steadily with age. These medications are commonly used to manage chronic conditions that become more prevalent as people get older, such as hypertension, diabetes, heart disease, arthritis, high cholesterol, and depression.
Generic drugs account for the large majority of prescriptions filled for older adults—about 90% of all retail or mail pharmacy prescriptions in the United States are dispensed as generics.
Generics contain the same active ingredients and must meet the same FDA standards for safety, quality, and efficacy as brand-name drugs, but typically cost much less. For seniors, the widespread availability and affordability of generics supports medication adherence and helps limit out-of-pocket costs, both for individuals and for public programs such as Medicare.
Because most major categories of chronic disease medications have generic options available, the majority of what seniors take each day is likely to be a generic product. This allows older adults to better manage multiple conditions as their needs increase with age, with important implications for quality of life and long-term health outcomes.
It is therefore fitting that the Senate Aging Committee is considering how best to assure that the generics seniors rely on remain safe, effective, and available. I appreciate the Committee’s attention to this issue and the opportunity to testify on steps we can take to strengthen those assurances.
My testimony proceeds in three steps. First, it explains how intense price competition shapes today’s generic markets and why those economics contribute to quality lapses and shortages. Second, it evaluates commonly proposed solutions through a fit-for-purpose and budget-conscious lens, highlighting where onshoring helps and where upstream China risks and hospital shortages merit higher priority than Indian finished dosage forms (FDF). Third, it turns to direct, scalable measures to strengthen Current Good Manufacturing Practice (CGMP) oversight—concluding with a Qualified Person (QP) certification model and targeted import testing to improve detection, deterrence, and accountability without destabilizing supply.
The race to the bottom
Generic drugs are prescription drugs that contain the same active ingredients, dosage form, strength, safety, quality, and intended use as their brand-name counterparts, but are typically sold at a significantly lower price than the brand they copy once sold at. In this section, I describe what drives this competition.
To be approved, generic drugs follow an abbreviated pathway that allows applicants to rely on existing clinical data from the brand-name product to establish safety and efficacy. The abbreviated pathway does not, however, waive manufacturing quality requirements: applicants must still prove to the FDA that their manufacturing processes can consistently replicate the approved product, ensuring each batch matches the quality, safety, and efficacy of the original drug.
While minor differences such as inactive ingredients, appearance, and packaging are permitted, generic drugs are regarded as interchangeable with their brand-name originals. This means that pharmacists can substitute a generic drug for the brand-name version without needing a new prescription, and patients can expect equivalent safety, efficacy, and clinical effects from either product. By extension, pharmacists may substitute one generic version for another.
Leveraging the therapeutic equivalence of generic versions, payers structure reimbursement systems—including Maximum Allowable Cost (MAC) lists, discounts off Wholesale Acquisition Cost (WAC), Average Sales Price (ASP) formulas, and Diagnosis Related Group (DRG) payments—to promote the selection of the least expensive option. In all these systems, providers and pharmacies are strongly incentivized to seek out the lowest-cost product that meets therapeutic equivalence requirements because reimbursement typically does not vary with the acquisition cost of different generic versions.
In the retail pharmacy setting, payers often reimburse pharmacies based on MAC lists, which set a fixed upper limit (the maximum allowable cost) for what the payer or pharmacy benefit manager will pay for a given generic drug, regardless of which manufacturer is chosen. Pharmacies may also be reimbursed using payment formulas (such as discounts off WAC) or other fixed amounts. In all cases, selecting a lower-cost generic increases the pharmacy’s margin, since reimbursement is not tied to the actual purchase price.
In inpatient hospital settings, commercial and government payers frequently bundle input costs into a single payment determined by DRG, so hospitals have a strong incentive to minimize input costs—including drugs—by using the least expensive generic version. For drugs administered in outpatient clinics, reimbursement may be bundled or based on ASP across all versions of the same generic drug. In this scenario, the payer’s outlay is not adjusted for which generic is sourced, again driving providers to select the lowest-priced option.
To obtain the best prices, major retail pharmacy chains participate in one of the four buying groups that represent over 90% of generic drug volume purchased. The four main joint ventures are: Red Oak Sourcing (CVS Health and Cardinal Health), Walgreens Boots Alliance (Walgreens and Cencora), ClarusOne (Walmart and McKesson), and Econdisc Contracting Solutions (Express Scripts and Kroger, among others). These joint ventures consolidate the purchasing power of their members, enabling them to negotiate deeper discounts and more favorable terms.
Similarly, hospitals pool their bargaining power using group purchasing organizations (GPOs). The top three GPOs collectively represent hospitals that account for over 80% of hospital beds, giving them substantial collective purchasing power to negotiate prices. Vizient holds the largest share of the market with 37% of hospital beds, followed by Premier with 28%, and HealthTrust with 15%.
Empirical data confirms continued price pressure resulting from concentrated purchasing power among retail chains and hospital networks.
There is continued deflation in the generic retail markets. Industry analyses report annual deflation rates of -10% to -15% for most generic drugs from 2017 to 2018, moderating somewhat to -5% to -10% by 2021. One analysis of pharmacy acquisition costs for top-selling generics found that many now cost pharmacies less than $1.50 for a 30-day supply. Most recently, a 2024 analysis found year-over-year acquisition cost deflation for generic oral solids as high as 25%—an unprecedented drop likely amplified by recent National Average Drug Acquisition Cost (NADAC) survey methodology changes. But recent manufacturing earnings reports reference continued price erosion, especially for older generic products.
Generic drug prices in hospital markets are also at historic lows. Analyses of FDA and US Pharmacopeia (USP) shortage databases show that over half of sterile injectables and 66% of solid oral medicines in shortage were invoiced at the pharmacy level for less than $5 and $3, respectively, in 2024. A recent IQVIA report found that while only 1% of drugs invoiced at $500 or more are in shortage, 11% of drugs priced under $1 are in shortage. As provided for this testimony by QuickSortRx Inc., among the 20 most commonly used generic injectables, eight had options with list prices below $1, another with options listed between $1 and $1.50, and only four had list prices of at least $5. In addition to list prices, some generics may also be providing GPO discounts or 340-B discounts. One 2018 analysis listed those in the range of discounted about 15% to 20% for GPOs and 25% to 50% for 340B. Those discounts have since had 7 more years to deepen.
Generic drug prices in hospital markets are also at historic lows. Analyses of FDA and USP shortage databases show that over half of sterile injectables and 66% of solid oral medicines in shortage were invoiced below $5 and $3, respectively, in 2024. A recent IQVIA report found that only 1% of drugs invoiced at $500 or more are in shortage, compared to 11% priced under $1. As provided for this testimony by QuickSortRx Inc., among the 20 most commonly used generic injectables, eight had list prices below $1, one ranged from $1 to $1.50, and only four were priced at $5 or more. Generics also often receive GPO or 340B discounts, which a 2018 analysis estimated at about 15% to 20% for GPOs and 25% to 50% for 340B—discounts that have likely grown deeper since.
The race has consequences
The high leverage of drug buyers leads to demand instability, which, when coupled with persistent price pressure, leads to significant pressure to cut costs. This can then lead to reduced supply chain resilience and may compromise the ability or willingness of manufacturers to maintain strict CGMP standards. Both can then lead to drug shortages. This section explores these operational realities and their consequences.
Demand instability
In the retail generics market, manufacturers often lack predictability over how much product they can sell while at least breaking even on production costs. Long-term contracts exist but frequently include best price guarantees that force manufacturers to match uncontracted competitors’ discounts—sometimes weekly and without knowing the quantities sold at those lower prices. In the hospital setting, similar clauses exist in GPO contracts but are triggered less often, likely because fewer manufacturers produce a given generic sterile injectable. However, hospitals can still source off-contract, further reducing the predictability of order volume.
Unstable demand makes production planning difficult for generic drug manufacturers. With lead times for raw materials and manufacturing often measured in months, companies must choose between producing larger, more efficient batches—risking unsold inventory if demand falls—or smaller, more costly ones to avoid losses.
The large product portfolios, coupled with uncertain demand, also create manufacturing control challenges. Unlike a branded production line, which may be dedicated to the same product for several years in a row, generic production lines may switch between 20 to 30 products in a year. For generic products first to market, batch runs may last a couple of months, but will shorten to three to ten days once the market settles and competitors emerge. These frequent changeovers not only increase labor and equipment downtime but also require rigorous regulatory documentation and validation, adding further complexity and cost to manufacturing operations. But if not conducted properly, changeovers can lead to cross-contamination between products.
Generic relative shock size
Faced with price pressure and unpredictable demand, manufacturers of generic drugs have a strong incentive to cut costs. Two of the ways that manufacturers can cut costs directly affect shock size: create scale and offshore. These are important from a supply resilience perspective, and therefore the risk of shortages, because the greater the shock relative to the market, the harder it is for supply chains to recover.
As described in the unstable demand section, manufacturers have a strong incentive to leverage economies of scale to minimize per-unit cost. However, the lack of predictability over demand tampers with those incentives. But if market structure allows for it, for example, sterile injectable facilities have high entry barriers and fewer competitors, scale wins. It is therefore why we see sterile injectable markets with single facilities representing 60% market share of 1L saline bags, 50% market share of injectable morphine, and a 50% market share of carboplatin.
Another market structure factor that may create disproportionately large shocks is the co-location of facilities. Economic incentives have a lot to do with it—moving facilities to lower-cost environments. For example, tax policies implemented in the 1950s encouraged significant expansion of the U.S. pharmaceutical industry in Puerto Rico. When Hurricane Maria hit the island in September 2017, most, if not all, of the 50 pharmaceutical facilities on the island were affected. Similarly, the low cost of labor and capital in East Asia, coupled with government subsidies in some of these countries, has shifted parts of pharmaceutical production to those countries, creating a potential geopolitical threat.
The impact of offshoring FDF production is clearly visible in Figure 1: 61% of the FDF of generic solid oral drugs sold in pharmacies are now primarily made in India. The U.S. makes 22% unit share of generic solid oral dose drugs, a big share of which are controlled substances such as opioids and ADHD medications, due to Drug Enforcement Agency requirements that those products are domestically produced. China represents a 3.5% unit share, which is less than Europe’s 5.4% unit share.
Sterile injectable generics, such as IV antibiotics, saline, chemotherapy agents, lidocaine, and epinephrine, are still largely made in the U.S. (Figure 1). Part of it has to do with the complexity of production relative to oral dose products and the much higher transportation costs for such drugs. But offshoring has followed, with India taking a leading role in the trend, currently at 20.9% unit share. China and Europe are almost on par when it comes to generic injectables, 11.8% and 12.7%, respectively.
Figure 1: Unit volume share of generic finished dosage drug production (2024)
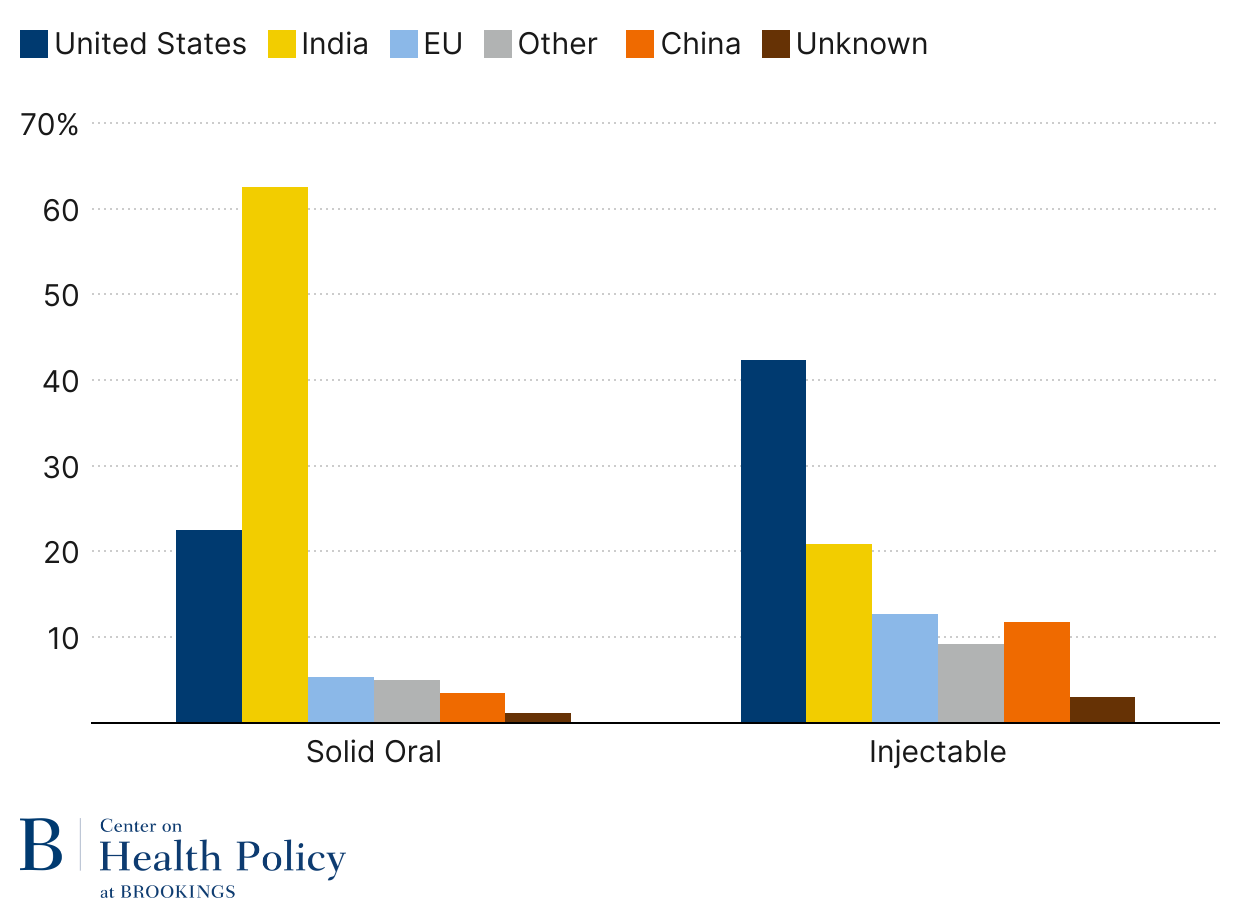
Source: Wosińska (2025), “Will pharmaceutical tariffs achieve their goals?“ using USP Medicine Supply Map.
Note: Solid oral dose volume is measured in tablets/capsules; injectables are measured in dispensing units (vials, syringes, or IV bags)
The upstream supply chains for pharmaceuticals—especially for active pharmaceutical ingredients (APIs), key starting materials, and certain bulk chemicals—have even greater exposure to international risk, with significant dependencies on India for active ingredients and on China for chemical precursors. However, when considering the physical product quality concern at the heart of this analysis, it is the FDF production stage that matters most. This is because CGMP compliance requires that manufacturers thoroughly test and document the quality of all inputs, regardless of where those inputs originate. If manufacturers are meeting their CGMP obligations, any product defects reaching the market, even if they originate with inputs, represent a breakdown at the final production stage.
Diminished ability to respond to shocks
When supply chain shocks occur, shortages result if the supply chains lack resilience to absorb or recover from them. If recovery is slow, shortages persist.
Branded drug manufacturers pay attention to supply chain resilience because high margins earned by their products provide manufacturers with strong incentives to invest in trying to prevent disruptions to those products. The risk of foregoing these margins and profits gives them a greater incentive to invest in quality systems and to maintain spare capacity in case production unexpectedly must shut down. When production disruptions of this kind occur, they tend to resolve faster.
For generics, low margins create little incentive to invest in resilience efforts, but actual resilience across generic drug markets differs because of market structure and technological differences.
Generic injectables often require specialized production lines (you cannot make one-liter IV bags on a line that only makes 250-milliliter IV bags). In other cases, it is because production lines are often dedicated to specific products (as with cancer drugs and antibiotics), as process requirements make switching lines either costly or impossible. Ramping up production can take weeks, even on a production line fitted for the drug. The market concentration described in the previous subsection also means that the gap to recover from can be substantial, e.g., half the market production that has ceased overnight.
Generic drugs that are formulated as tablets or capsules face possibly greater price pressures from pharmacies, but they are less vulnerable to shortages because they face a different manufacturing environment and market structure. Manufacturing of those products is less complex, not requiring specialized facilities with employees following complex manufacturing processes and controls. Generic oral dose product markets are also less concentrated than sterile injectable ones, and there is more fungibility in oral dose production lines that can also be ramped up faster.
However, common shocks can still be quite consequential in the oral solid dose market, despite these advantages. For example, when many manufacturing facilities are co-located in a single region, they may all be vulnerable to the same natural disaster or local infrastructure failure. Similarly, reliance on common sources for raw materials or key ingredients, or siting production in countries that may impose export bans, can create systemic supply risks. Such vulnerabilities mean that even the relatively resilient retail generic market is not immune to wide-ranging disruptions.
While geographic concentration and upstream supply chains are important risks, it is important to note that much of the fragility in the generic drug supply chain is associated with manufacturing quality failures in the production of the FDF (i.e., tablet or vial of medicine). This, in turn, is closely associated with adherence to CGMPs.
Challenges to upholding CGMPs
High-quality, safe, and effective generic drugs depend on robust compliance with CGMPs. CGMPs are essential safeguards, ensuring that medicines meet strict standards for identity, strength, quality, and purity in every batch produced, minimizing risks of contamination, error, or deviation. Because neither patients nor clinicians can readily assess the quality, safety, or efficacy of medicines themselves, the public relies on manufacturers to maintain these rigorous standards and on FDA to enforce them through an established regulatory framework.
Manufacturers bear primary responsibility for following CGMPs, which require oversight at every step of production—monitoring equipment, verifying staff credentials, supervising processes, testing samples, and thoroughly investigating any problems that arise. This is especially critical for complex products like sterile injectables, where lapses in quality control can result in serious contamination risks.
Yet, significant price competition and unstable demand often pressure manufacturers to cut costs in ways that undermine CGMP compliance. Thin margins and unpredictable markets can lead to reductions in Quality Unit staffing, less independence for those responsible for quality assurance, and the blending of production and quality roles. Manufacturers may also limit routine controls, such as audits and investigations, weakening the systemic safeguards needed to ensure each product’s safety and reliability.
As pharmaceutical supply chains grow more global and complex, FDA’s oversight is more important—and more challenged—than ever. The agency must hold manufacturers accountable for consistently upholding these essential practices in the face of ongoing operational and economic pressures.
FDA enforcement faces headwinds
To ensure consistent drug quality, FDA relies on a combination of prescriptive regulatory requirements, facility inspections, and various compliance tools. FDA requires manufacturers to have strong quality systems that follow CGMPs, including oversight of every production step, checking samples during production, and keeping thorough records. FDA investigators then verify compliance with standards through both scheduled and unannounced inspections, during which they review everything from personnel training and equipment maintenance to contamination control and recordkeeping. When serious violations are uncovered, FDA can act through warning letters, drug recalls, import refusals, and import alerts.
But the FDA oversight system is imperfect. FDA struggles with persistent staffing shortages and inspection backlogs, especially for foreign facilities in countries such as India and China. Many inspections abroad are still announced in advance, which weakens the detection of compliance issues. Driven by economic forces, some pharmaceutical manufacturers of low-cost generics often have weak quality management systems that lack the authority and resources to enforce CGMP compliance effectively. Perhaps most concerning, decisions not to release a batch or to initiate a recall are often judgment calls. For low-margin products, manufacturers face strong incentives to release a questionable product rather than bear the financial hit of withholding it from distribution.
The consequences of manufacturing quality problems can require costly remediation, but shortages also invite government support. Here is what might be referred to as a “too important to fail” problem: FDA does not want shortages, so it will be flexible with its rules when problems arise, especially for medically necessary drugs where the manufacturer, in violation of FDA standards, also holds a large market share. That dynamic, however, undermines the deterrence effect of fines, just like a traffic cop giving out warning tickets for speeding will have a lesser deterrence impact than a cop giving out fines.
In such an environment, manufacturers may function at a “lowest common denominator” level of compliance, where CGMP principles are not fully enforced, and quality systems lack the independence or resources to reliably detect, investigate, and prevent errors.
Analyses of FDA enforcement trends and warning letters identify recurring failures in quality unit authority, documentation practices, change control, and batch record review—especially among overseas and generic manufacturers. FDA data show that in recent years, many facilities inspected—particularly in China, India, and Southeast Asia—received citations for inadequate quality management, data integrity lapses, and insufficient oversight of outsourced production. Systemic noncompliance at these facilities increases the risk that substandard or contaminated drugs will reach patients, with potentially serious health consequences.
At the same time, the detection of such quality oversight problems can force manufacturers or regulators to delay or withhold release of affected products. This restriction—while essential to protect patients—often leads directly to drug shortages. Manufacturing quality issues are a leading cause, responsible for 56% in 2011, 62% between 2013 and 2017, and 46% in 2022 of shortage events.
The illusion of easy fixes
Policy solutions must be fit for purpose. Measures that don’t match the failure they target risk being inadequate—or even counterproductive—in a market defined by intense price competition and thin margins.
For supply chains, there is one overarching goal: ensuring patients can access safe and effective drugs when needed. To achieve this, it is important to distinguish four related but distinct concepts:
- Physical product quality: reducing product defects in the drug itself, including contamination, mislabeling, and potency failures.
- Quality assurance: strengthening systems that prevent defects—manufacturing controls, process capability, data integrity, and ongoing monitoring—because unit-by-unit verification is infeasible and CGMP is the principal assurance tool.
- Supply chain resilience: improving the ability to withstand disruptions—manufacturing problems, input shortages, plant closures, or geopolitical shocks—regardless of origin.
- Supply chain reliability: the broadest outcome, encompassing both quality assurance and resilience, to deliver dependable, on-time availability of medicines.
In this section, I evaluate policy options to address manufacturing quality concerns identified by this Committee, assessing them on how well they advance each of the four concepts outlined above, with attention to whether the costs are justified by the benefits for patients and taxpayers.
Transparency for consumers
At the September 17th hearing, patient-facing transparency around drug quality drew genuine interest from several senators, who were intrigued by its potential for empowering consumers and strengthening trust in the pharmaceutical system. The underlying idea is straightforward: studies show consumers would value drug quality information, and if prescription labels included measures like a five-star rating based on product testing, patients could express preferences at the pharmacy, potentially steering demand toward products with higher ratings.
However, what this idea overlooks is that pharmacies have little economic incentive to accommodate such requests. Pharmacies are reimbursed the same amount regardless of the manufacturer’s quality rating, meaning they earn no more for dispensing a higher-rated product. In fact, sourcing a non-preferred or specially ordered product often carries additional costs, including time, administrative burden, and inventory risk, without any offsetting revenue—a challenge for what is already a low-margin business.
Patients, meanwhile, have only one real choice at the pharmacy counter, which is to take the medication offered or forgo treatment altogether. This means the only way to express preference in the current system is through non-adherence—a harmful and unacceptable outcome.
Even if drug quality scores were printed on labels, pharmacies would be unlikely to change procurement patterns unless reimbursement models tied payments to quality ratings, making it economically viable to stock or special-order higher-rated products. Without correcting this misalignment, consumer-driven quality improvement could make things worse, not better. Policymakers should therefore proceed carefully with the idea, either by addressing pharmacy reimbursement first or by turning to other tools to accomplish the same goal of improved drug quality of drugs sold in retail pharmacies.
That is not to say transparency is all bad. For example, with greater visibility into supply chains, the federal government can assess which supply chains are most vulnerable, prioritizing those for intervention. Quality metrics can also be critically important for institutional buyers—an issue I turn to next.
Transparency for institutional buyers
Unlike individual patients, institutional buyers such as hospital pharmacies, mail-order, and retail pharmacies have real choices when it comes to generic manufacturers from whom they procure. This makes transparency around supply reliability and manufacturer quality an especially valuable area to explore.
The early development and application of these ideas have focused heavily on the hospital setting, where drug shortages have been particularly common and acute.
The central goal of these transparency initiatives is to equip institutional buyers with metrics that help them identify manufacturers capable of reliably delivering products. This focus is distinct from typical product quality measures; it is about shifting demand toward manufacturers with stronger records of supply chain reliability. By doing so, the overall stability and resilience of the pharmaceutical supply chain can be strengthened as demand incentivizes better performance.
Efforts to create such metrics are not new. As early as 2012, the concept was first proposed in a research paper I coauthored with then-FDA Drug Center Director Janet Woodcock. This work has since evolved into three key programs aimed at measuring and reporting on manufacturer quality and supply reliability, including the FDA Quality Management Maturity (QMM) assessments, Healthcare Industry Resilience Collaborative (HIRC), and US Pharmacopeia’s (USP) quality testing and resiliency benchmarking programs. All these remain in early or pilot phases. Their eventual adoption, implementation, and influence on the market are not yet certain. Nevertheless, their shared aspiration is to drive market incentives toward a more robust and reliable drug supply.
Addressing this gap, some entities like Civica Rx operate with a different model, vetting manufacturers rigorously before entering into long-term supply agreements, thereby providing member hospitals with a dependable drug supply as well as encouraging manufacturers to maintain high reliability standards.
Ultimately, transparency about supply reliability alone will not transform institutional purchasing behavior unless accompanied by changes to the underlying incentives faced by hospitals and GPOs. Even now, GPOs could use their size and collective purchasing power to assess supply chain vulnerabilities, including by compelling confidential information about manufacturers’ supply reliability. But GPOs are only paid if hospitals buy through their contract, and with hospitals seeking the best prices, neither GPOs nor hospitals have sufficient incentive to pursue such initiatives aggressively.
In other words, no matter how much transparency there is about suppliers, if there is no financial reward or accountability on the hospital’s part for not having a drug available, transparency reforms will falter.
This discussion, therefore, turns to the bipartisan Senate Finance proposal put forward in May 2024, which aims to reform the hospital incentives to select more reliable manufacturers.
Senate Finance proposal
Because the Senate Finance legislative drug shortage proposal addresses the fundamental incentives hospitals face, it stands as perhaps the most consequential attempt to correct the persistent equilibrium of manufacturing quality issues that trigger a disproportionate share of shortages.
The Senate Finance Committee’s approach focuses on generic sterile injectable drugs where the shortage risk is the greatest. The proposal has two pillars.
First, the legislative proposal provides for Medicare add-on payments to hospitals and physicians who sign long-term contracts for qualifying generic sterile injectables with more reliable supply chains. The proposal would require providers and GPOs to sign minimum three-year contracts, bind themselves to meaningful purchase volumes, and maintain contingency agreements with alternate manufacturers. The premise is that such stable purchasing will help ensure continuous drug supply and encourage manufacturers to invest in quality and capacity, knowing they will have committed buyers.
The program’s success hinges on the practicality of participation costs for hospitals and having a reliable way to identify which manufacturers are trustworthy, meaning that they can consistently deliver drugs that meet specifications to hospitals. Efforts to develop quality metrics for reliability are important, but an alternative approach is simply to pay based on whether the contracted drug was delivered to specification, sidestepping the challenge of metric precision.
The second major component is reforming the ability of generic manufacturers to increase prices. Current Medicaid policies—particularly the inflationary rebate system—cap how much manufacturers can pass costs onto the Medicaid program. The same rebates extend to the 340B, in which about half the hospitals participate. The inflation rebate, while designed to prevent price gouging, caps all price increases, even when those increases could fund investment in better quality or redundancy. To address that, the Senate Finance proposal eliminates Medicaid inflation rebates and, by extension, addresses the same problem in the 340B program.
Since the proposal has come out, many stakeholders have raised concerns that adjusting the inflation rebate and 340B interactions would harm hospital finances. But for shortage-prone generics, the dollars at issue are small not only to overall hospital pharmacy budgets but also the intake from the branded-side of the 340B program. It is also not realistic to fund multi-year quality investments by ‘raising price elsewhere.’ Attempting to pass the full share of quality improvements to non-340B buyers is likely to run up against what is effectively a price-cap that is an add-on payment, risking loss of participation by those buyers if the potentially doubly higher prices would not be worth the add-on.
Overall, the Senate Finance proposal is a promising framework, but it requires significant refinement to offer true relief from drug shortages without creating new inefficiencies. It addresses the market most vulnerable to shortages, the generic sterile injectable market. This leaves the retail market concerns still to be addressed.
Domestic manufacturing
Onshoring production could indeed help improve CGMP compliance by lowering the rate of product defects for generics. One benefit of shifting production domestically is the likelihood of building new facilities that incorporate more advanced manufacturing methods and process automation. Such upgrades can lower the risk of human error and increase operational consistency, reducing the potential for product defects. Modern systems also enable better and faster detection of problems through real-time monitoring, robust documentation, and integrated quality management protocols. FDA faces fewer barriers to robust facility oversight within the U.S., further reinforcing CGMP compliance.
But a reality check is needed: using domestic manufacturing as a systemic fix for CGMP shortcomings runs straight up against powerful economic headwinds. The offshoring of generic drug production has been shaped by the market forces I described earlier. Reversing this trend and building up domestic supply chains at scale would require a major government intervention—meaning sustained new spending on subsidies, direct investment, or tax credits to counteract those underlying economic pressures.
Recent data highlight the immensity of the foreign footprint. As Figure 1 indicates, India supplied 61% of over 187 billion generic pills dispensed in U.S. retail and mail pharmacies in 2024. This volume came from a combination of 179 sites in India making FDF drugs. In addition, up to 236 Indian sites supplied APIs. China’s role in FDF generics is smaller, with 60 facilities paying generic facility fees in 2024, but it dominates further upstream, including 148 API sites and a large presence in critical starting materials, reagents, solvents, and intermediates used across the pharmaceutical manufacturing chain (see Wosinska 2025).
Replicating this fully onshore would require hundreds of billions of dollars of new construction, supplemented by ongoing operational subsidies to overcome persistent cost, scale, and policy disadvantages vis-à-vis India and China (see Wosinska 2025)—the same factors that drove manufacturing out of the U.S.
If Congress authorizes less than what is required to fully onshore supply chains, including the chemicals that supply the pharmaceutical production, policymakers will face difficult trade-offs in allocating limited funds. They will need to weigh risks such as persistent drug shortages, dependence on a small number of foreign suppliers for APIs and chemical inputs, vulnerability to geopolitical disruptions, and the challenge of scaling U.S. manufacturing capacity efficiently and promptly.
Government onshoring initiatives should explicitly prioritize the highest-risk segments first—or risk spending heavily and achieving little improvement in supply reliability. Without clear prioritization under tight budgets, funds will diffuse across too many targets, yielding marginal resilience gains while wasting taxpayer and patient dollars. Policymakers should therefore concentrate investments on essential APIs, lifesaving hospital injectables prone to recurring shortages, and upstream chemical inputs with the greatest China-dependence, sequencing other categories only after these choke points are secured.
Generic FDFs sourced from India—such as common oral tablets like statins and antihypertensives, produced at massive scale—are not at the top of the onshoring priority list. For this reason, we should look for alternative solutions that directly address persistent CGMP shortcomings in Indian facilities.
Tariffs
Before turning to alternative solutions to CGMP concerns in Indian (and Chinese) facilities, it is important to discuss tariffs as an onshoring tool because of the unintended consequences that tariffs may have on generic drug supply availability and quality.
The implied mechanism for how tariffs lead to onshoring begins with tariffs increasing the prices that foreign firms can charge, making their products less competitive in the U.S. market. As these prices rise, demand naturally shifts away from the now more expensive foreign suppliers toward existing domestic manufacturers. This shift allows domestic firms not only to gain market share but also to raise their own prices, thereby increasing their profitability. As domestic firms become more profitable, they are incentivized to expand production capacity, and the improved business environment attracts new manufacturers to establish operations in the U.S.
This mechanism, however, requires that prices can adjust, with it shifting demand. This, however, is limited in the case of generic drugs by several key market and regulatory features. Medicaid inflation rebates create a cap on how much manufacturers can increase prices, with any additional price increases rebated to Medicaid. Additionally, the 340B program—whose discounts are pegged to Medicaid prices—extends these caps to outpatient drugs used by about half the hospitals. Beyond outpatient 340B use, hospitals may also use contracts that cap for one to three years, preventing price hikes even when input costs rise.
But with margins low, the inability to recover now higher costs means manufacturers affected by tariffs will face a choice: either further cut costs or exit the market.
The potential for further cutting costs is concerning because it can adversely affect product quality if cost-cutting happens through equipment maintenance, quality of materials, process control, or quality assurance. If problems arise, for example, the product is contaminated with other API, bacteria, or metal shavings (all actual examples), manufacturers may temporarily shut down or slow down production, leading to a potentially dramatic drop in output. But if FDA oversight is concurrently weakened, economic theory and experience suggest we should expect product quality to decrease.
Another bad option is to discontinue production of the unprofitable drug. Historically, discontinuations have not been a major driver of shortages, partly because manufacturers have tended to decrease production before exiting, leaving a more vulnerable market but not triggering a shortage. But the optics might change here – companies may be less shy about exiting the market with a higher market share due to tariffs they cannot control. There is also a concern that discontinuations of smaller share markets can occur in close succession, combining the impact of each.
This disruption in supply would not be as problematic if the share of the affected market is small or if the affected markets do not have substantial exposure to either the Medicaid program or the hospital market. For example, Chinese products are already facing a 20% tariff, but no shortages have resulted. This is likely a combination of the relatively small presence of Chinese products in the U.S. market (3.5% for oral dose and 11.8% for injectables, per Figure 1), the types of drugs that Chinese manufacturers are making and therefore the exposure those drugs have to price caps, and the margins that those manufacturers might have (including through potential Chinese government subsidies).
However, depending on the size of the tariff, a tariff on Indian pharmaceuticals plays out differently because many markets would face little alternative to Indian supplies. In particular, there is limited ready-to-activate domestic manufacturing capacity to quickly replace lost production. However, onshoring generic drug production faces substantial obstacles. Building or expanding pharmaceutical manufacturing in the U.S. involves high capital costs, reaching hundreds of millions of dollars per facility, and construction plus regulatory approvals can take three to five years or more.
Without substantive payment reform, generic drug margins will continue to be thin. Even with tariffs, the incremental gain from domestic production is unlikely to offset these substantial costs. Firms and investors remain hesitant to pursue capital-intensive investments, given persistent uncertainty about the duration of tariff protection—especially considering President Trump’s recent use of pharmaceutical tariffs as a threat for concessions against branded manufacturers rather than a predictable policy tool.
Margins and market exposure may vary across products, but the evidence is clear that unless pass-through of tariff costs is addressed, higher tariffs on generics will increase the risk of shortages in markets with Medicaid or 340B exposure—particularly if a large enough share of the market is affected by these tariffs.
Strengthening FDA enforcement
Over the years, various proposals have been put forward to address the gap in oversight between foreign and domestic facilities, which by law had to be inspected every two years, in contrast to foreign facilities, which had no such requirement.
After two decades of faltering foreign inspections, the onset of the Generic Drug User Fees Amendments (GDUFA) provided a much-needed infusion of funding for foreign inspections. However, FDA continued to struggle with leveling the playing field between domestic and foreign facilities, especially those located in India and China, partly because GDUFA also fueled the number of generic drug approvals, which in turn increase demand for foreign inspections.
Since the onset of GDUFA, FDA and outside observers such as the Government Accountability Office (GAO) have called for a number of changes to FDA’s foreign facility oversight in countries without comparable regulatory oversight and therefore within Mutual Recognition Agreements (MRA) with FDA. Recommendations include expanding unannounced inspections, increasing inspection frequency, improving policies to ensure inspector independence, addressing inspection workforce and backlog challenges, and exploring alternative monitoring tools, such as remote access to documents, information from foreign regulators’ inspections, or third-party audits.
Due to time constraints in preparing this testimony, I am not reviewing the specific proposals or initiatives individually. What can be said is that each one of them strengthens the probability of detecting CGMP problems. FDA has been acting on those recommendations, but they all collectively require additional resources and, therefore, support from Congress. But it is important to say that they focus on increased detection, without changing any rules of the game when it comes to accountability. I turn to that concept next.
Raising the bar on accountability
An alternative but complementary path to enhance U.S. drug quality assurance involves requiring every importer to designate a qualified individual—located in the U.S.—who certifies that each batch meets CGMP standards, and to ensure that mandatory testing of imported products is also conducted in the U.S. This system already exists in the EU for drugs and has precedent under the Food Safety Modernization Act, specifically with the Foreign Supplier Verification Program (FSVP) and Preventive Controls rules, which each designate a “Qualified Individual” or a “Preventive Controls Qualified Individual” to oversee compliance activities.
Under the Qualified Person (QP) system, each EU importer designates a QP with specialized credentials and formal authority to personally certify that every drug batch meets GMP and regulatory requirements before release. The QP reviews all relevant batch data, including manufacturing and quality control records, and ensures compliance with the marketing authorization. They may rely on audits or testing within the EU, but bear personal legal responsibility for certification.
For drug imports from countries lacking a Mutual Recognition Agreement (MRA) with the EU, the QP must, before certifying any batch, also conduct mandatory re-testing of each batch at EU-registered, GMP-compliant laboratories. This further ensures product quality and reduces reliance on the exporting country’s regulatory system.
Layering a QP system onto FDA’s existing system would change both the probability of problems being detected and the consequences once they are uncovered. Detection probability would increase through required import testing and the additional review conducted by QPs, whether through documentation checks or site audits. Consequences would increase because—in addition to the legal responsibility of the firm—the QP would now be personally responsible for certifying each batch and, therefore, unwilling to approve products whose CGMP compliance is in doubt. Conversations with current and past QPs confirm that dynamic.
The QP system, paired with targeted import testing, directly addresses the underlying CGMP challenge: ensuring that only medicines meeting robust manufacturing standards can enter the U.S. supply chain. This approach is more efficient and focused than indirect levers like tariffs or wholesale onshoring, which may impose far greater costs but do not directly improve product quality assurance. Compared to indirect levers like tariffs or broad onshoring, the QP testing approach provides a direct, efficient, and scalable path to enhanced product quality assurance.
In the broader policy context, the QP testing approach is distinct from, but works alongside, other interventions such as the Senate Finance proposal’s hospital reliability incentives. The QP testing model is not a replacement for supply-focused solutions: rather, it fills a critical gap in product-level quality assurance, especially for generic drugs in the retail channel, where reliability incentives cannot prevent substandard products from reaching patients. Senate Finance’s proposal largely addresses injectable hospital drugs but does not reach the retail segment or guarantee batch-level manufacturing quality.
Well-designed drug quality policy should target known vulnerabilities rather than assume risk is limited to foreign or non-U.S. facilities. Accountability mechanisms like the QP testing proposal must therefore be tailored—fit for purpose, nuanced, and proportional—so that the approach delivers both detection and deterrence, and focuses responsibility precisely where oversight is weakest, and consequences for U.S. patients are greatest.
Implementing a QP certification and mandatory batch testing system in the U.S. raises several practical considerations, discussed in detail in the accompanying QP report. Key design decisions include determining the scope of products subject to the requirements (e.g., imports from non-MRA countries), establishing standards for QP certification and liability protections, accrediting U.S. testing labs, and phasing in requirements to prevent supply disruptions. Policymakers should also account for reimbursement barriers in Medicaid, 340B, and other programs, and ensure cost and regulatory burdens remain proportional to the policy’s risk-targeting aims. Attention to these design, resourcing, and rollout challenges will be critical to fully realizing the benefits of accountability-focused reform.
Conclusion
Patients deserve safe, high-quality medicines, and companies are legally required to deliver them by upholding robust CGMP standards. Yet, economic pressures and FDA’s expanding oversight constraints have created a system that too often fails to deliver on that requirement. Policymakers are right to seek solutions.
But in identifying solutions, policymakers must be careful that policy interventions are fit for purpose—that is, tailored to the specific problem and designed to achieve the intended outcome without creating new risks or costs. Put simply, the tool should match the failure it aims to fix, not a broader goal that drifts from the underlying cause.
If the goal is simply to increase domestic drug manufacturing, almost any facility will qualify regardless of clinical need or shortage risk, and little attention may be paid to whether tools to drive onshoring could destabilize supply. If China is the central concern, attention should turn upstream—to the raw materials, reagents, and chemical inputs where supply chains are most exposed—while India should be leveraged as a strategic partner to de-risk from China at lower cost than direct U.S. production. If persistent hospital drug shortages are the true priority, durable solutions require shifting hospital incentives, so reliability is valued more than low cost.
Beyond being fit for purpose, policy solutions must also account for costs and budget constraints. With finite resources and competing risks, not every problem can or should be addressed through onshoring; under realistic budgets, Indian FDFs are unlikely to be top priorities compared to essential APIs, hospital injectables prone to shortage, and upstream chemicals with concentrated China exposure. Responsible policy should pursue strategies that deliver the best value not only for patients but also for taxpayers.
For concerns about defective drugs reaching patients from non-MRA countries, the most direct and cost-effective approach is to improve manufacturing quality oversight. Many proposals understandably focus on increasing the probability of detecting problems through expanded and unannounced inspections, stronger overseas enforcement, and increased testing, but accountability has received less attention despite being equally critical.
Introducing a QP system, complemented by mandatory import testing of every batch, provides a stronger safeguard for drug quality. Combining mandatory product testing of imports with batch certification by a qualified U.S.-based person who bears personal sign-off responsibility strengthens both detection and deterrence and underscores that safeguarding drug quality and CGMP compliance is an industry obligation—not a task that can be offloaded to regulators alone.
Related Content

Author

-
Acknowledgements and disclosures
This testimony draws on work conducted with support from the Gary and Mary West Health Policy Center.
The Brookings Institution is committed to quality, independence, and impact.
We are supported by a diverse array of funders. In line with our values and policies, each Brookings publication represents the sole views of its author(s).


Commentary
TestimonyWhen cheap becomes fragile: How the race to the bottom in generics undermines manufacturing quality and what to do about it
March 24, 2026